 Получите свидетельство
Получите свидетельство Вход
Вход




Факторы риска возникновении наследственных заболеваний человека Мастер-класс
Детский клуб « MEL » Санкт-Петербург
Руководитель проекта Павлова Вероника Дмитриевна
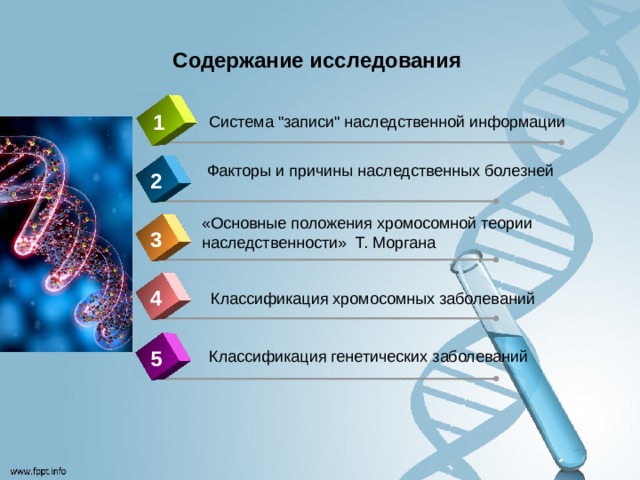
Содержание исследования
1
Система "записи" наследственной информации
Факторы и причины наследственных болезней
2
«Основные положения хромосомной теории наследственности» Т. Моргана
3
4
Классификация хромосомных заболеваний
5
Классификация генетических заболеваний
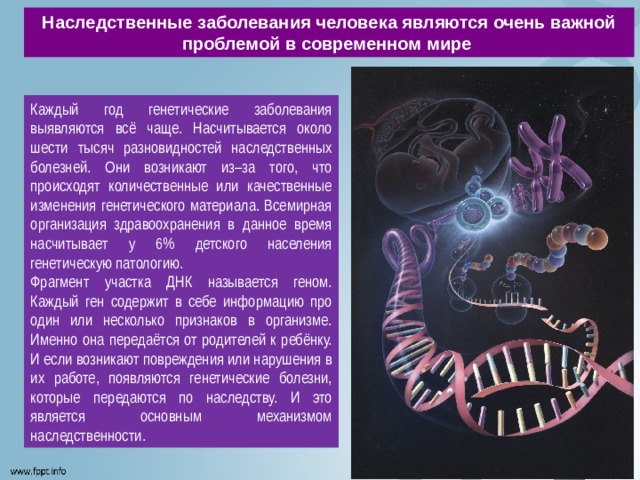
Наследственные заболевания человека являются очень важной проблемой в современном мире
Каждый год генетические заболевания выявляются всё чаще. Насчитывается около шести тысяч разновидностей наследственных болезней. Они возникают из–за того, что происходят количественные или качественные изменения генетического материала. Всемирная организация здравоохранения в данное время насчитывает у 6% детского населения генетическую патологию.
Фрагмент участка ДНК называется геном. Каждый ген содержит в себе информацию про один или несколько признаков в организме. Именно она передаётся от родителей к ребёнку. И если возникают повреждения или нарушения в их работе, появляются генетические болезни, которые передаются по наследству. И это является основным механизмом наследственности.
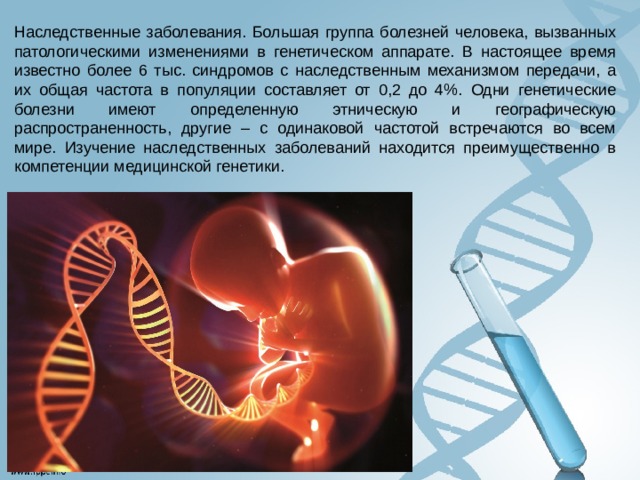
Наследственные заболевания. Большая группа болезней человека, вызванных патологическими изменениями в генетическом аппарате. В настоящее время известно более 6 тыс. синдромов с наследственным механизмом передачи, а их общая частота в популяции составляет от 0,2 до 4%. Одни генетические болезни имеют определенную этническую и географическую распространенность, другие – с одинаковой частотой встречаются во всем мире. Изучение наследственных заболеваний находится преимущественно в компетенции медицинской генетики.
Организм человека является сложной системой
Заложенный в организме генетический код управляет развитием зародыша, позволяет сформироваться всем взаимодействиям и обуславливает нормальное существование человека.
В наследственной информации могут появиться ошибки, возникшие на уровне отдельных генов или же их крупных объединений. Подобные изменения называются генными мутациями.
Проблемы возникшие в структурных единицах клетки хромосомах, называются хромосомными мутациями.
Сцепленное с полом наследование – это наследование признаков, гены которых локализованы в половых хромосомах - открытие Т. Морганом. Признаки. У человека признаки, наследуемые через Y- хромосому , могут быть только у лиц мужского пола, а наследуемые через X- хромосому - у лиц обоих полов.
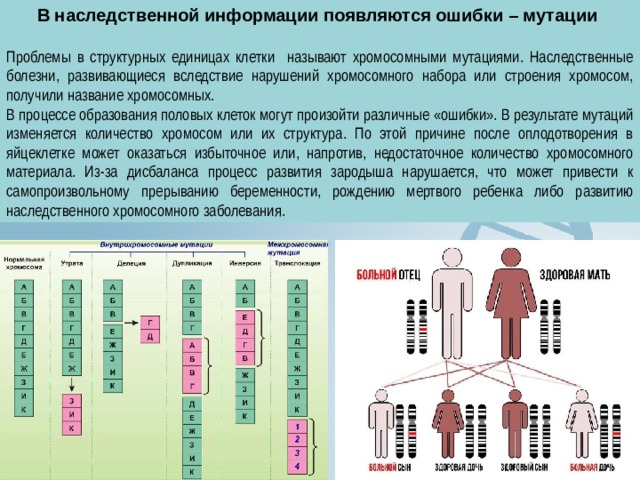
В наследственной информации появляются ошибки – мутации
Проблемы в структурных единицах клетки называют хромосомными мутациями. Наследственные болезни, развивающиеся вследствие нарушений хромосомного набора или строения хромосом, получили название хромосомных.
В процессе образования половых клеток могут произойти различные «ошибки». В результате мутаций изменяется количество хромосом или их структура. По этой причине после оплодотворения в яйцеклетке может оказаться избыточное или, напротив, недостаточное количество хромосомного материала. Из-за дисбаланса процесс развития зародыша нарушается, что может привести к самопроизвольному прерыванию беременности, рождению мертвого ребенка либо развитию наследственного хромосомного заболевания.
Факторы риска возникновении наследственных заболеваний
Наследственность – свойство организмов сохранять и обеспечивать передачу наследственных признаков потомкам, а также программировать особенности их индивидуального развития в конкретных условиях среды. Нормальные и патологические признаки организма являются результатом взаимодействия наследственных (внутренних) и средовых (внешних) факторов.
Одним из методов дородовой (пренатальной) диагностики плода является амниоцентез. Данная процедура заключается в заборе околоплодных вод c дальнейшим проведением их гормонального, биохимического, цитологического (исследование хромосомного состава клеток плода) и иммунологического анализа.

Томас Хант Морган (1866 – 1945) стал лауреатом Нобелевской премии в области физиологии и медицины в 1933 году, сформулировал
«Основные положения хромосомной теории наследственности»
Его хромосомная теория объяснила природу наследственных патологий человека, позволила экспериментально изменять наследственную информацию и стала началом современных методов генетических исследований. Не будучи первооткрывателем.
Томас Хант Морган сформулировал постулаты теории, которая изменила мир.
Томас Хант Морган Выдающийся учёный исследователь оставил после себя ряд научных работ, давших толчок в развитии, генетики.
Важнейшими из них считают:
«Регенерация» (1901г.),
«Экспериментальная зоология»,
«Наследственность и пол» (1913г.),
«Критика теории эволюции», «Физические основы наследственности» (1932г.),
«Генетика дрозофильной мухи» «Теория гена» (1932г.).

У человека не может быть искусственного скрещивания
- в силу его биосоциальности;
- низкая плодовитость;
- редкая смена поколений;
- наличие в геноме большого
числа групп сцепления;
- высокая степень
фенотипического полиморфизма.
Эти особенности не дают возможность применять на человеке гибридологический метод исследования Георга Менделя
Классификация мутаций
По характеру возникновения
По локализации в организме
Спонтанные, индуцированные
По локализации в клетке
Генеративные, соматические
По проявлению
Ядерные, цитоплазматические
По уровню изменения генотипа
Доминантные, рецессивные
Геномные, генные, хромосомные
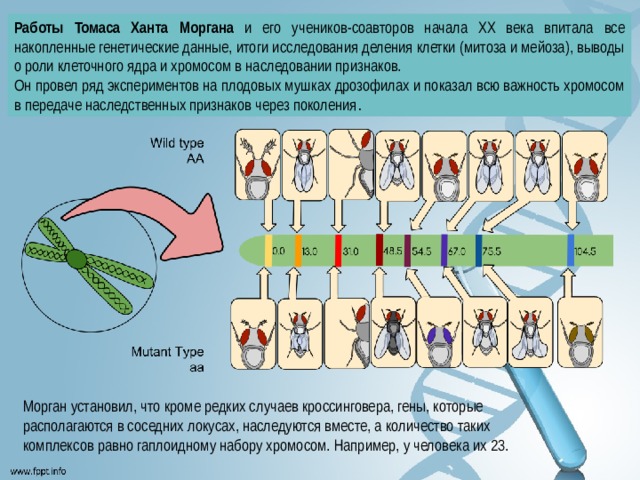
Работы Томаса Ханта Моргана и его учеников-соавторов начала XX века впитала все накопленные генетические данные, итоги исследования деления клетки (митоза и мейоза), выводы о роли клеточного ядра и хромосом в наследовании признаков.
Он провел ряд экспериментов на плодовых мушках дрозофилах и показал всю важность хромосом в передаче наследственных признаков через поколения .
Морган установил, что кроме редких случаев кроссинговера, гены, которые располагаются в соседних локусах, наследуются вместе, а количество таких комплексов равно гаплоидному набору хромосом. Например, у человека их 23.
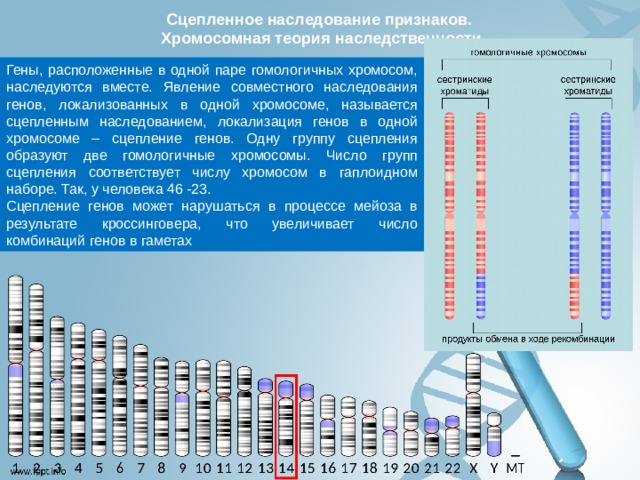
Сцепленное наследование признаков. Хромосомная теория наследственности
Гены, расположенные в одной паре гомологичных хромосом, наследуются вместе. Явление совместного наследования генов, локализованных в одной хромосоме, называется сцепленным наследованием, локализация генов в одной хромосоме – сцепление генов. Одну группу сцепления образуют две гомологичные хромосомы. Число групп сцепления соответствует числу хромосом в гаплоидном наборе. Так, у человека 46 -23.
Сцепление генов может нарушаться в процессе мейоза в результате кроссинговера, что увеличивает число комбинаций генов в гаметах
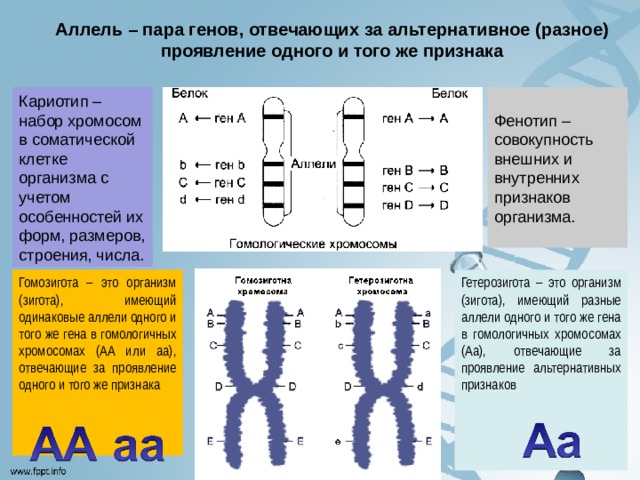
Аллель – пара генов, отвечающих за альтернативное (разное) проявление одного и того же признака
Кариотип – набор хромосом в соматической клетке организма с учетом особенностей их форм, размеров, строения, числа.
Фенотип – совокупность внешних и внутренних признаков организма.
Гетерозигота – это организм (зигота), имеющий разные аллели одного и того же гена в гомологичных хромосомах (Аа), отвечающие за проявление альтернативных признаков
Гомозигота – это организм (зигота), имеющий одинаковые аллели одного и того же гена в гомологичных хромосомах (АА или аа), отвечающие за проявление одного и того же признака
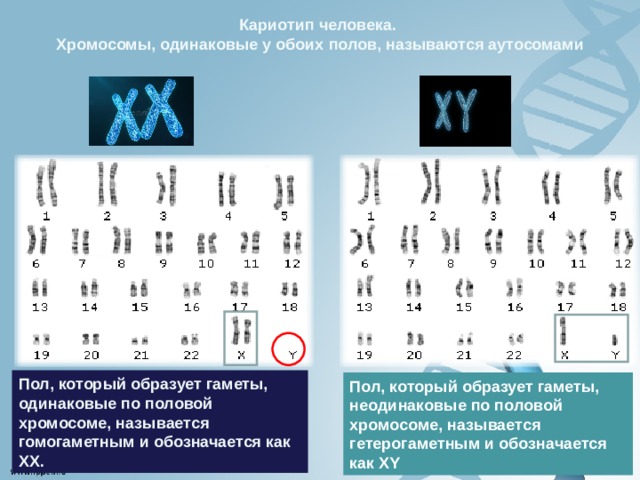
Кариотип человека.
Хромосомы, одинаковые у обоих полов, называются аутосомами
Пол, который образует гаметы, одинаковые по половой хромосоме, называется гомогаметным и обозначается как XX.
Пол, который образует гаметы, неодинаковые по половой хромосоме, называется гетерогаметным и обозначается как XY
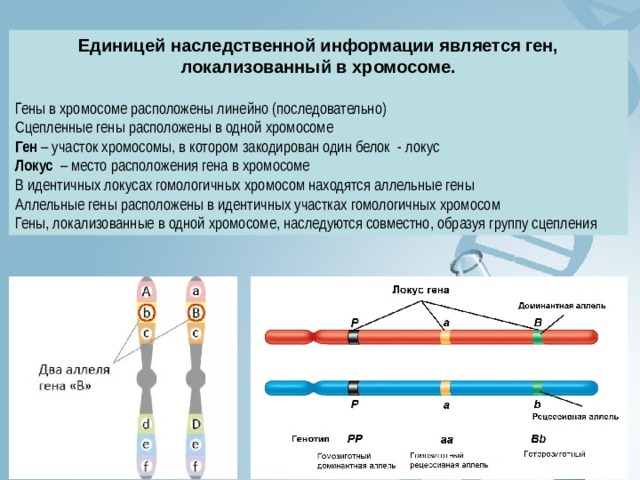
Единицей наследственной информации является ген, локализованный в хромосоме.
Гены в хромосоме расположены линейно (последовательно)
Сцепленные гены расположены в одной хромосоме
Ген – участок хромосомы, в котором закодирован один белок - локус
Локус – место расположения гена в хромосоме
В идентичных локусах гомологичных хромосом находятся аллельные гены
Аллельные гены расположены в идентичных участках гомологичных хромосом
Гены, локализованные в одной хромосоме, наследуются совместно, образуя группу сцепления
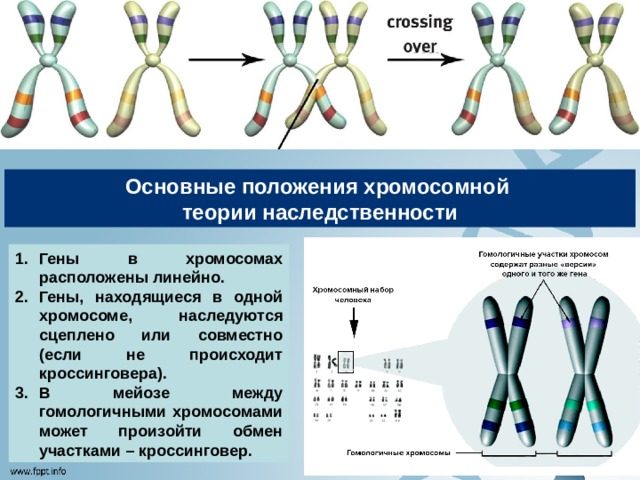
Основные положения хромосомной
теории наследственности
- Гены в хромосомах расположены линейно.
- Гены, находящиеся в одной хромосоме, наследуются сцеплено или совместно (если не происходит кроссинговера).
- В мейозе между гомологичными хромосомами может произойти обмен участками – кроссинговер.
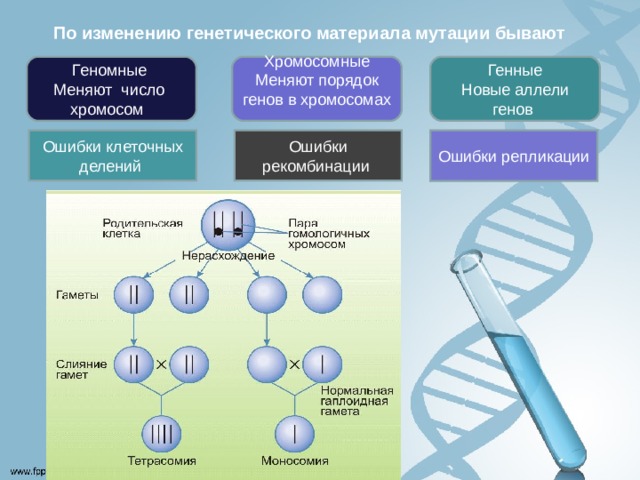
По изменению генетического материала мутации бывают
Генные
Новые аллели генов
Хромосомные Меняют порядок генов в хромосомах
Геномные
Меняют число хромосом
Ошибки репликации
Ошибки клеточных делений
Ошибки рекомбинации

Мутации являются естественным процессом и составляют основу эволюционной изменчивости всех живых существ
Признаки, определяемые генами, находящимися в Х – хромосомах, называются признаками, сцепленными с полом. Наследование таких признаков, было открыто и изучено на дрозофиле Т. Морганом. и послужило ярким доказательством локализации генов в хромосомах.
Возникновение генетических болезней происходит тогда, когда происходит мутация – нарушается механизм хранения и передачи генетического материала. При повреждении гена эта информация будет передаваться следующему поколению так же, как и материал, не подвергшийся мутации.
Причины
Моногенные
Болезни
Мутации или отсутствие гена в ядерной ДНК
Синдром Марфана, адреногенитальный синдром у новорожденных, нейрофиброматоз, гемофилия А, миопатия Дюшенна
Полигенные
Предрасположенность и действие экзогенных факторов
Псориаз, шизофрения, ишемическая болезнь, цирроз, бронхиальная астма, сахарный диабет

Геномные мутации – изменение числа хромосом
(нарушение мейоза)
Полиплодии – кратное увеличение полного набора хромосом
2n + n 2n + 2n 2n + 3n …
Анеуплодии – изменение числа хромосом в одной или нескольких парах . Изменение числа хромосом в кариотипе. - кратное геному изменение числа хромосом
2n + 1 2n + 2 … 2n – 1 2n – 2 …
- Триплодия
- Тетраплодия
- Моносомия
- Трисомия
У человека несовместимы с жизнью
Синдром Дауна - трисомия по 21 хромосоме, кариотип – 47 хромосом.
Синдром Эдвардса - трисомия по 18 хромосоме
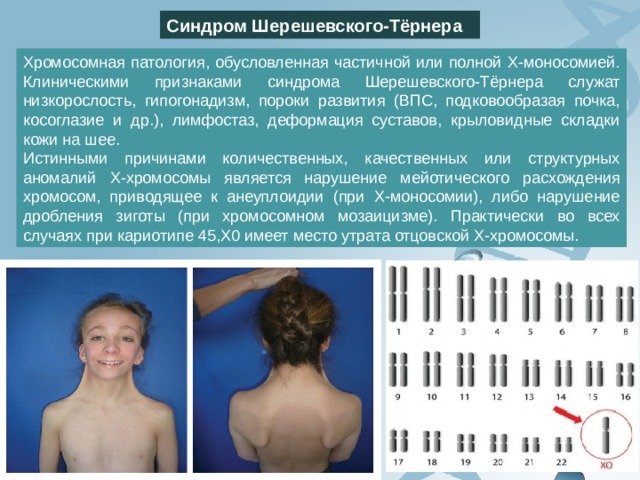
Синдром Шерешевского-Тёрнера
Хромосомная патология, обусловленная частичной или полной Х-моносомией. Клиническими признаками синдрома Шерешевского-Тёрнера служат низкорослость, гипогонадизм, пороки развития (ВПС, подковообразая почка, косоглазие и др.), лимфостаз, деформация суставов, крыловидные складки кожи на шее.
Истинными причинами количественных, качественных или структурных аномалий Х-хромосомы является нарушение мейотического расхождения хромосом, приводящее к анеуплоидии (при Х-моносомии), либо нарушение дробления зиготы (при хромосомном мозаицизме). Практически во всех случаях при кариотипе 45,Х0 имеет место утрата отцовской Х-хромосомы.
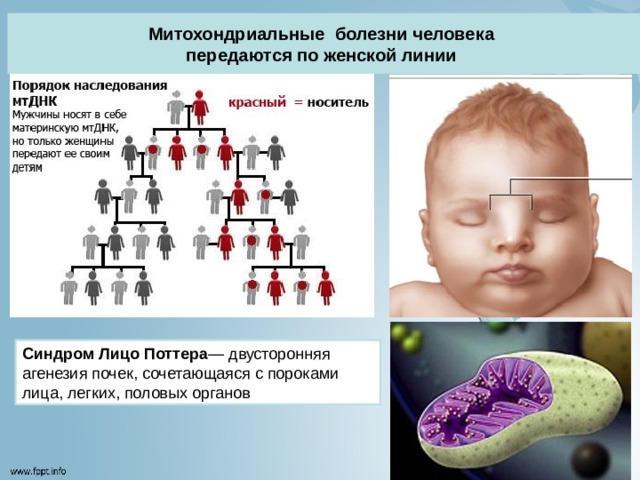
Митохондриальные болезни человека
передаются по женской линии
Синдром Лицо Поттера — двусторонняя агенезия почек, сочетающаяся с пороками лица, легких, половых органов

Хромосомные болезни
Хромосомные болезни, или синдромы - это группа врожденных патологических состояний, проявляющихся множественными пороками развития, различающихся по своей клинической картине, часто сопровождающихся тяжелыми нарушениями психического и соматического развития.
Основной дефект - различные степени интеллектуальной недостаточности, что может осложняться нарушениями зрения, слуха, опорно-двигательного аппарата, более выраженными, чем интеллектуальный дефект, расстройствами речи, эмоциональной сферы и поведения.
Хромосомные заболевания не подчиняются менделеевским закономерностям передачи заболевания потомству и в большинстве случаев являются следствием мутации в половой клетке одного из родителей.
- проявляются с первых дней жизни;
- задержка общего физического и психического развития;
- черепнолицевые аномалии, аномалии других частей скелета;
- грубые пороки сердечнососудистой, мочеполовой и нервной системы, отклонения в биохимическом, гормональ ном, иммунном статусе;
- малая продолжительность жизни.
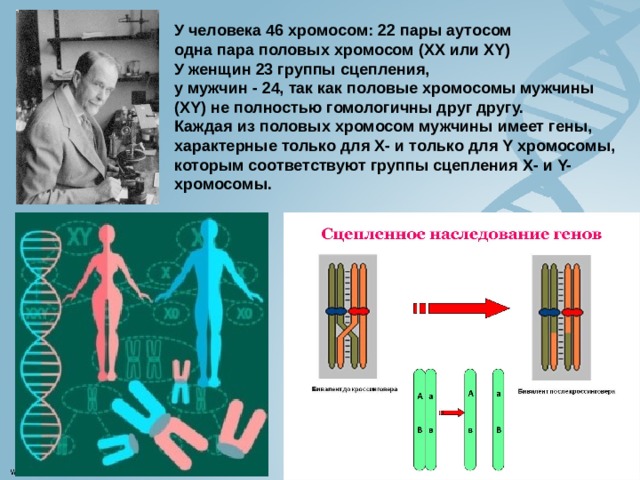
У человека 46 хромосом: 22 пары аутосом
одна пара половых хромосом (XX или XY) У женщин 23 группы сцепления,
у мужчин - 24, так как половые хромосомы мужчины (XY) не полностью гомологичны друг другу. Каждая из половых хромосом мужчины имеет гены, характерные только для Х- и только для Y хромосомы, которым соответствуют группы сцепления Х- и Y-хромосомы.
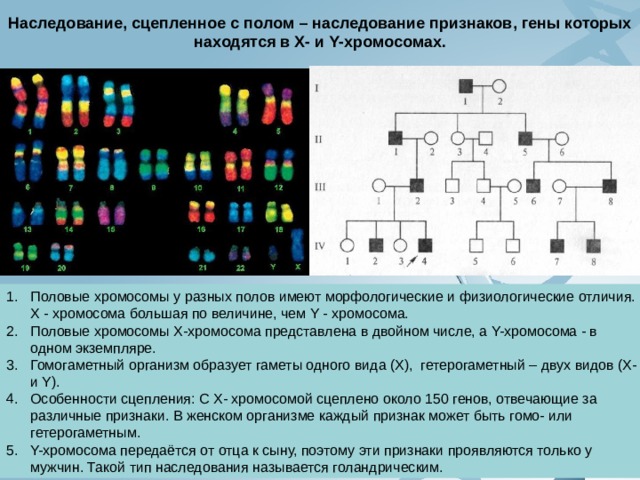
Наследование, сцепленное с полом – наследование признаков, гены которых находятся в Х- и Y-хромосомах.
- Половые хромосомы у разных полов имеют морфологические и физиологические отличия. Х - хромосома большая по величине, чем Y - хромосома.
- Половые хромосомы Х-хромосома представлена в двойном числе, а Y-хромосома - в одном экземпляре.
- Гомогаметный организм образует гаметы одного вида (Х), гетерогаметный – двух видов (Х-и Y).
- Особенности сцепления: С Х- хромосомой сцеплено около 150 генов, отвечающие за различные признаки. В женском организме каждый признак может быть гомо- или гетерогаметным.
- Y-хромосома передаётся от отца к сыну, поэтому эти признаки проявляются только у мужчин. Такой тип наследования называется голандрическим.
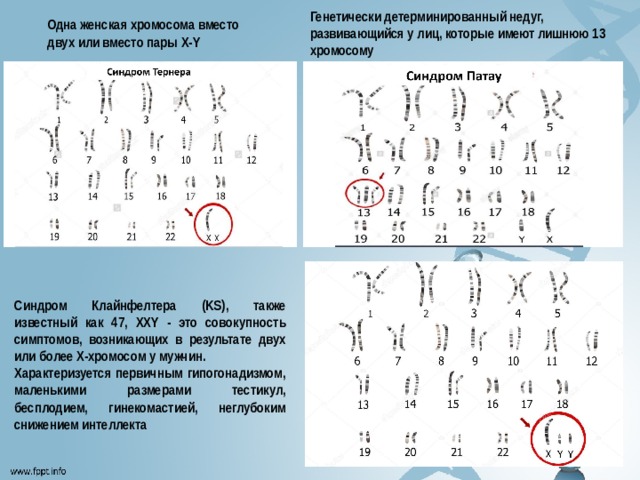
Генетически детерминированный недуг, развивающийся у лиц, которые имеют лишнюю 13 хромосому
Одна женская хромосома вместо двух или вместо пары Х-Y
Синдром Клайнфелтера (KS), также известный как 47, XXY - это совокупность симптомов, возникающих в результате двух или более Х-хромосом у мужчин.
Характеризуется первичным гипогонадизмом, маленькими размерами тестикул, бесплодием, гинекомастией, неглубоким снижением интеллекта
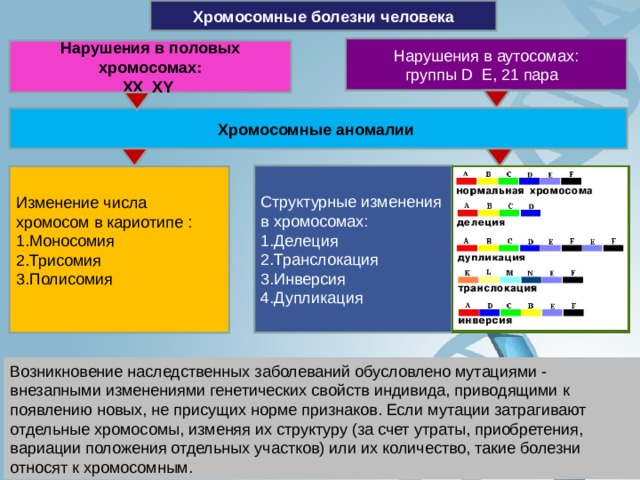
Хромосомные болезни человека
Нарушения в аутосомах:
группы D E , 21 пара
Нарушения в половых хромосомах:
XX XY
Хромосомные аномалии
Структурные изменения в хромосомах:
- Делеция
- Транслокация
- Инверсия
- Дупликация
Изменение числа хромосом в кариотипе :
- Моносомия
- Трисомия
- Полисомия
Возникновение наследственных заболеваний обусловлено мутациями - внезапными изменениями генетических свойств индивида, приводящими к появлению новых, не присущих норме признаков. Если мутации затрагивают отдельные хромосомы, изменяя их структуру (за счет утраты, приобретения, вариации положения отдельных участков) или их количество, такие болезни относят к хромосомным.

Хромосомные заболевания связаны с аномалиями числа или структуры хромосом.
Хромосомные болезни – комплексы множественных врожденных пороков развития, вызываемых числовыми (геномные мутации) или структурными (хромосомные аберрации).
Хромосомные аберрации и изменения количества хромосом, могут возникать на разных этапах развития организма. Если они возникают в гаметах родителей, то аномалия будет наблюдаться во всех клетках развивающегося организма. Если аномалия возникает в процессе эмбрионального развития при дроблении зиготы, кариотип плода будет мозаичным. Решающим фактором в проявлении хромосомного заболевания является возникновение в гаметах или зиготе на первых этапах её дробления хромосомного нарушения.

Синдром Мартина-Белла - это патология ломкой
X- хромосомы.
Ребенок имеет ярко выраженные отклонения в области психического и соматического состояния.
Синдром «кошачьего крика»
– хромосомное нарушение, обусловленное делецией (отсутствием) фрагмента короткого плеча 5-ой хромосомы.
Синдром Вольф-Хирсхорна (потеря короткого плеча 4-й) Внутриутробная гипоплазия, микроцефалия, гипертелоризм, широкий нос, ассимметрия черепа, низко расположенные уши, пороки сердца и почек у 50%, задержка умственного развития
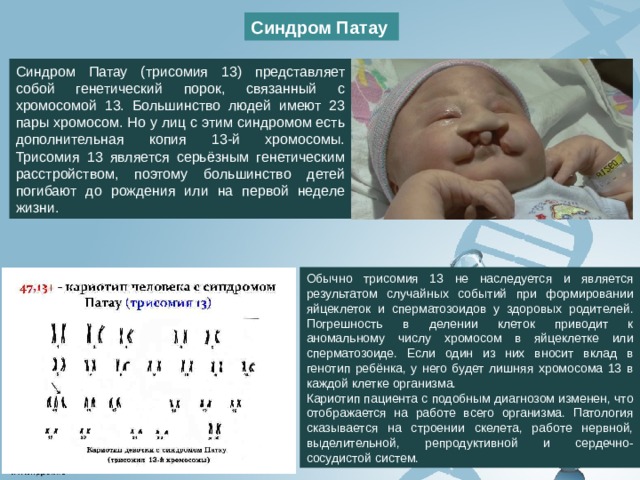
Синдром Патау
Синдром Патау (трисомия 13) представляет собой генетический порок, связанный с хромосомой 13. Большинство людей имеют 23 пары хромосом. Но у лиц с этим синдромом есть дополнительная копия 13-й хромосомы. Трисомия 13 является серьёзным генетическим расстройством, поэтому большинство детей погибают до рождения или на первой неделе жизни.
Обычно трисомия 13 не наследуется и является результатом случайных событий при формировании яйцеклеток и сперматозоидов у здоровых родителей. Погрешность в делении клеток приводит к аномальному числу хромосом в яйцеклетке или сперматозоиде. Если один из них вносит вклад в генотип ребёнка, у него будет лишняя хромосома 13 в каждой клетке организма.
Кариотип пациента с подобным диагнозом изменен, что отображается на работе всего организма. Патология сказывается на строении скелета, работе нервной, выделительной, репродуктивной и сердечно-сосудистой систем.
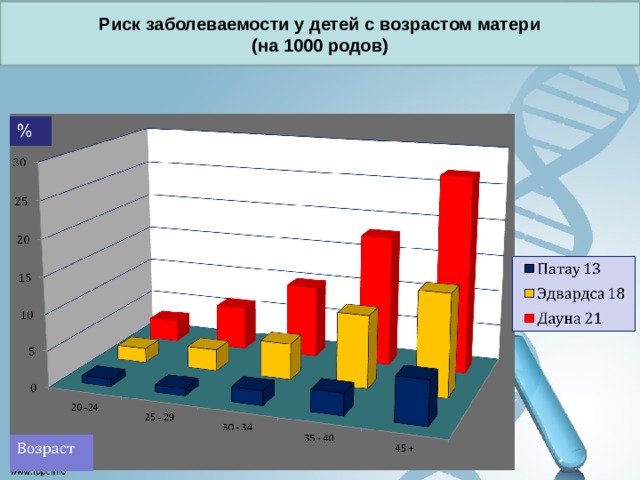
Риск заболеваемости у детей с возрастом матери
(на 1000 родов)
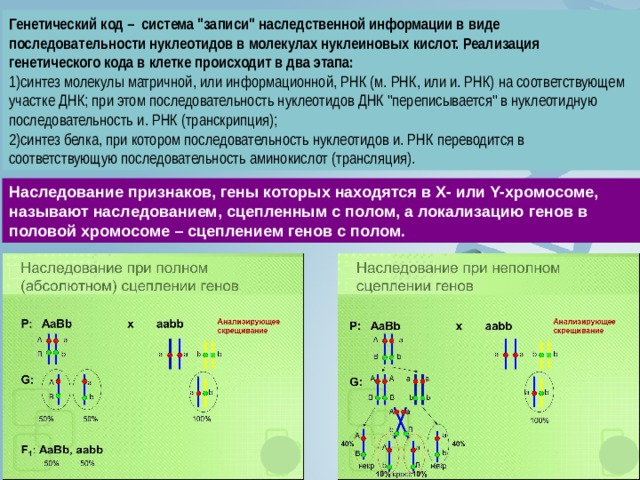
Генетический код – система "записи" наследственной информации в виде последовательности нуклеотидов в молекулах нуклеиновых кислот. Реализация генетического кода в клетке происходит в два этапа:
- синтез молекулы матричной, или информационной, РНК (м. РНК, или и. РНК) на соответствующем участке ДНК; при этом последовательность нуклеотидов ДНК "переписывается" в нуклеотидную последовательность и. РНК (транскрипция);
- синтез белка, при котором последовательность нуклеотидов и. РНК переводится в соответствующую последовательность аминокислот (трансляция).
Наследование признаков, гены которых находятся в Х- или Y-хромосоме, называют наследованием, сцепленным с полом, а локализацию генов в половой хромосоме – сцеплением генов с полом.

Сцепление генов может нарушаться в процессе мейоза в результате кроссинговера.
В процессе мейоза гомологичные хромосомы, а следовательно, и аллельные гены попадают в разные гаметы. Гаметы всегда гаплоидны. Негомологичные хромосомы, а следовательно и неаллельные гены расходятся произвольно, независимо друг от друга и образуют различные комбинации в гаметах
Генные болезни – это группа заболеваний, возникающих
в результате повреждений ДНК на уровне гена.
Генные мутации могут приводить:
- к синтезу аномального белка;
- к снижению количества синтезируемого белка;
- к увеличению количества синтезируемого белка;
- к отсутствию синтеза белка.
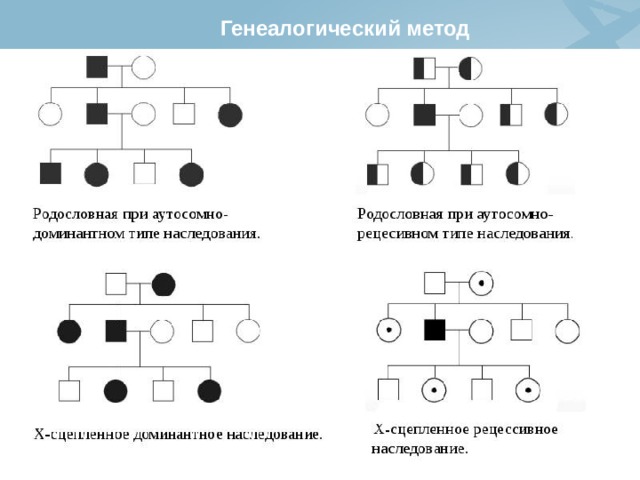
Генеалогический метод
относится к числу основных методов генетики человека и лежит в основе медико-генетического консультирования.
Генеалогический метод включает три этапа:
- сбор данных обо всех родственниках обследуемого (анамнез);
- составление родословных;
- генеалогический анализ родословных.

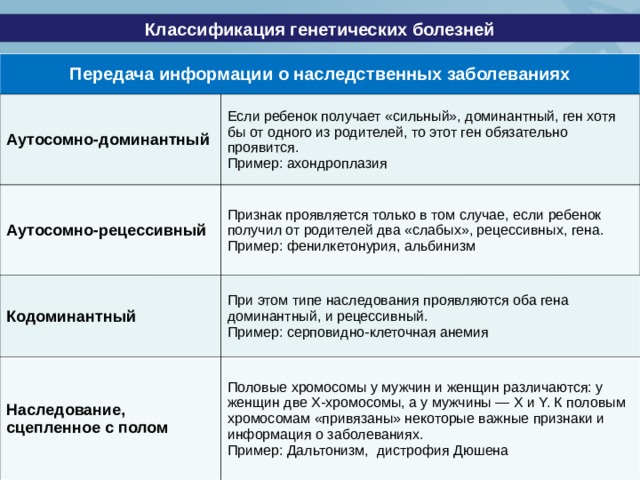
Классификация генетических болезней
Передача информации о наследственных заболеваниях
Аутосомно-доминантный
Если ребенок получает «сильный», доминантный, ген хотя бы от одного из родителей, то этот ген обязательно проявится.
Пример: ахондроплазия
Аутосомно-рецессивный
Признак проявляется только в том случае, если ребенок получил от родителей два «слабых», рецессивных, гена.
Пример: фенилкетонурия, альбинизм
Кодоминантный
При этом типе наследования проявляются оба гена доминантный, и рецессивный.
Пример: серповидно-клеточная анемия
Наследование, сцепленное с полом
Половые хромосомы у мужчин и женщин различаются: у женщин две Х-хромосомы, а у мужчины — X и Y. К половым хромосомам «привязаны» некоторые важные признаки и информация о заболеваниях.
Пример: Дальтонизм, дистрофия Дюшена
Генеалогический метод

Псевдогипопаратиреоз –
Болезнь Олбрайта
Болезнь Олбрайта - наследственная остеодистрофия, обусловленная резистентностью периферических тканей к паратгормону, что сопровождается расстройством кальциево-фосфорного обмена, задержкой физического и умственного развития.
Заболевание носит наследственный характер с аутосомно-доминантным типом наследования, сцепленным с X-хромосомой.
Изучение родословных показывает, что число женщин с псевдогипопаратиреозом в 2 раза превышает количество больных мужчин; кроме того, болезнь Олбрайта не передается от отца к сыновьям.
Псевдогипопаратиреоз обусловлен генетической резистентностью скелета и почек к действию паратиреоидного гормона.
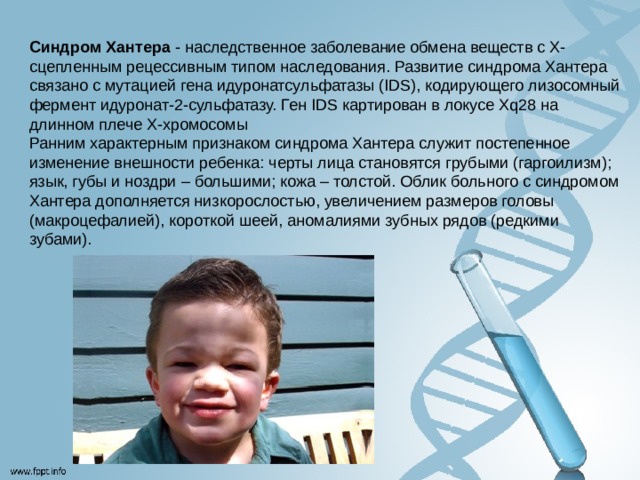
Синдром Хантера - наследственное заболевание обмена веществ с Х-сцепленным рецессивным типом наследования. Развитие синдрома Хантера связано с мутацией гена идуронатсульфатазы (IDS), кодирующего лизосомный фермент идуронат-2-сульфатазу. Ген IDS картирован в локусе Xq28 на длинном плече Х-хромосомы Ранним характерным признаком синдрома Хантера служит постепенное изменение внешности ребенка: черты лица становятся грубыми (гаргоилизм); язык, губы и ноздри – большими; кожа – толстой. Облик больного с синдромом Хантера дополняется низкорослостью, увеличением размеров головы (макроцефалией), короткой шеей, аномалиями зубных рядов (редкими зубами).
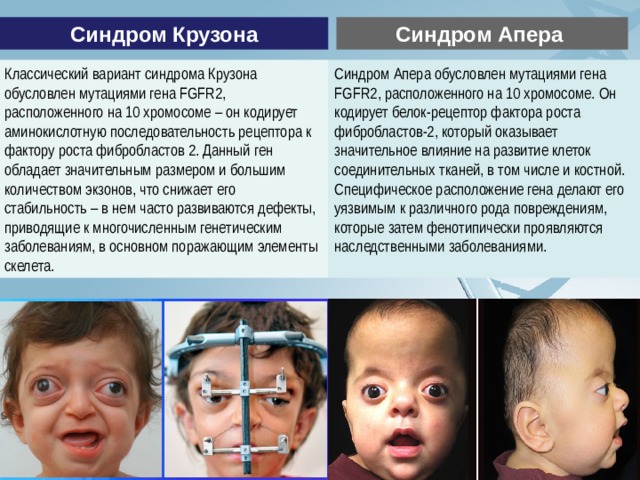
Синдром Крузона
Синдром Апера
Классический вариант синдрома Крузона обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме – он кодирует аминокислотную последовательность рецептора к фактору роста фибробластов 2. Данный ген обладает значительным размером и большим количеством экзонов, что снижает его стабильность – в нем часто развиваются дефекты, приводящие к многочисленным генетическим заболеваниям, в основном поражающим элементы скелета.
Синдром Апера обусловлен мутациями гена FGFR2, расположенного на 10 хромосоме. Он кодирует белок-рецептор фактора роста фибробластов-2, который оказывает значительное влияние на развитие клеток соединительных тканей, в том числе и костной. Специфическое расположение гена делают его уязвимым к различного рода повреждениям, которые затем фенотипически проявляются наследственными заболеваниями.
Несовершенный остеогенез
синдром Вролика-Лобштейна
«Хрустальные люди»
Фибродисплазия оссифицирующая прогрессирующая Мюнхеймера
«Каменные люди»
Нарушается структура коллагена, входящего в состав костей и других соединительных тканей, либо синтезируется его недостаточное количество.
Развитие болезни связано с врожденным нарушением обмена белка соединительной ткани коллагена 1-го типа, обусловленным мутациями генов, кодирующих коллагеновые цепи.
В зависимости от формы заболевание может наследоваться по аутосомно-доминантному или аутосомно-рецессивному типу (менее 5%). Примерно в половине случаев патология возникает вследствие спонтанных мутаций
ФОП возникает из–за мутации в гене ACVR1/ALK2, кодирующем рецептор костного морфогенетического белка. Редкое генетическое заболевание, при котором организм начинает формировать новые кости — оссификаты — в неположенных местах: внутри мышц, связок, сухожилий и других соединительных тканей. К их образованию может привести любая травма: ушиб, порез, перелом, внутримышечная инъекция или операция.. Физиологически оссификаты не отличаются от обыкновенных костей и могут выдерживать значительные нагрузки.

Ахондроплазия - болезнь Парро-Мари
Карликовость
Гипофизарная карликовость
(нанизм)
Аутосомно-доминантный тип наследования:
- больные встречаются в каждом поколении;
- болеют как мужчины, так и женщины
Гипофизарная карликовость (нанизм) – это заболевание, проявляющееся в задержке роста и физического развития ввиду нарушения секреции передней долей гипофиза соматотропина – гормона роста. Наибольшее распространение имеет пангипопитуитарная карликовость, которая имеет свойство наследоваться в основном по рецессивному типу.
Два типа формы передачи данной патологии – аутосомная и через Х-хромосому.
Является наиболее распространенной формой хондродисплазии и возникает из-за мутациив гене рецептора фактор роста фибробластов 3-го типа. Для больных характерны короткие руки и ноги, в то время как размеры туловища остаются нормальными или только немного меньше обычных. Как правило, у больных увеличена голова, а лоб выдается вперед. Рост взрослого человека с около 130 см
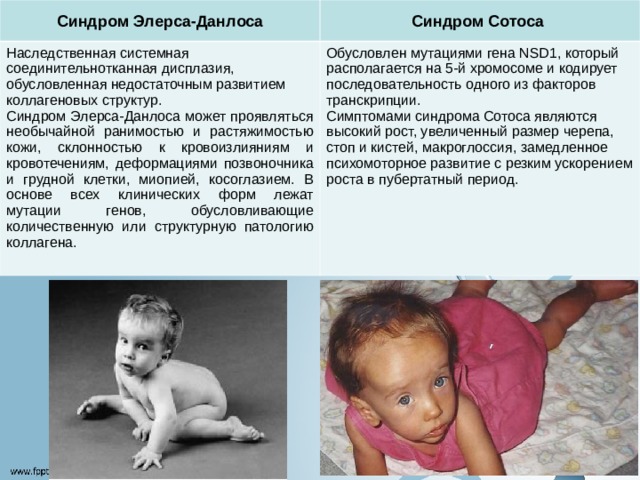
Синдром Элерса-Данлоса
Синдром Сотоса
Наследственная системная соединительнотканная дисплазия, обусловленная недостаточным развитием коллагеновых структур.
Синдром Элерса-Данлоса может проявляться необычайной ранимостью и растяжимостью кожи, склонностью к кровоизлияниям и кровотечениям, деформациями позвоночника и грудной клетки, миопией, косоглазием. В основе всех клинических форм лежат мутации генов, обусловливающие количественную или структурную патологию коллагена.
Обусловлен мутациями гена NSD1, который располагается на 5-й хромосоме и кодирует последовательность одного из факторов транскрипции. Симптомами синдрома Сотоса являются высокий рост, увеличенный размер черепа, стоп и кистей, макроглоссия, замедленное психомоторное развитие с резким ускорением роста в пубертатный период.
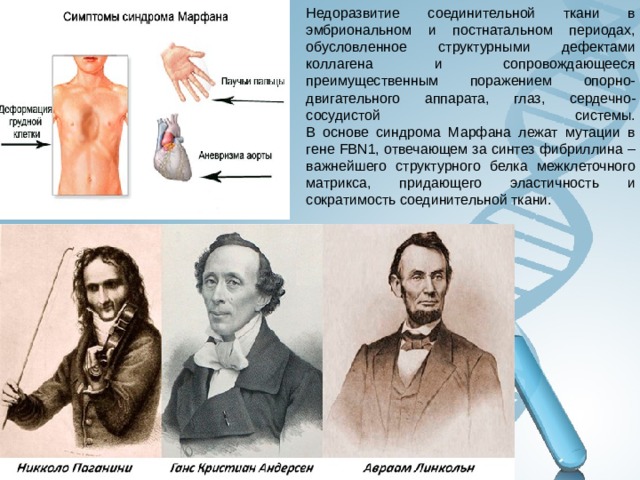
Недоразвитие соединительной ткани в эмбриональном и постнатальном периодах, обусловленное структурными дефектами коллагена и сопровождающееся преимущественным поражением опорно-двигательного аппарата, глаз, сердечно-сосудистой системы. В основе синдрома Марфана лежат мутации в гене FBN1, отвечающем за синтез фибриллина – важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани.
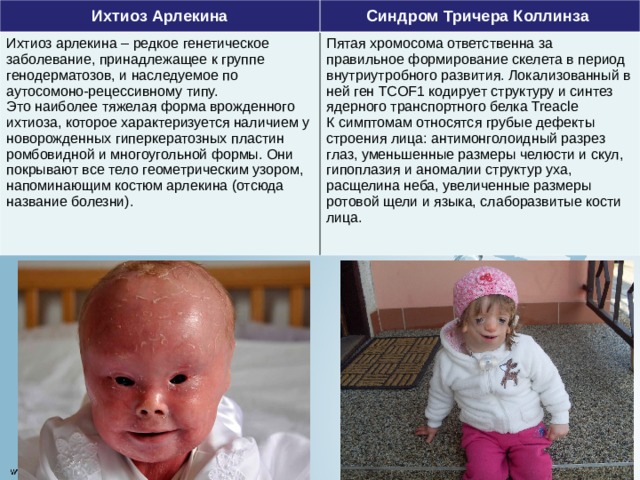
Ихтиоз Арлекина
Синдром Тричера Коллинза
Ихтиоз арлекина – редкое генетическое заболевание, принадлежащее к группе генодерматозов, и наследуемое по аутосомоно-рецессивному типу.
Это наиболее тяжелая форма врожденного ихтиоза, которое характеризуется наличием у новорожденных гиперкератозных пластин ромбовидной и многоугольной формы. Они покрывают все тело геометрическим узором, напоминающим костюм арлекина (отсюда название болезни).
Пятая хромосома ответственна за правильное формирование скелета в период внутриутробного развития. Локализованный в ней ген TCOF1 кодирует структуру и синтез ядерного транспортного белка Treacle К симптомам относятся грубые дефекты строения лица: антимонголоидный разрез глаз, уменьшенные размеры челюсти и скул, гипоплазия и аномалии структур уха, расщелина неба, увеличенные размеры ротовой щели и языка, слаборазвитые кости лица.
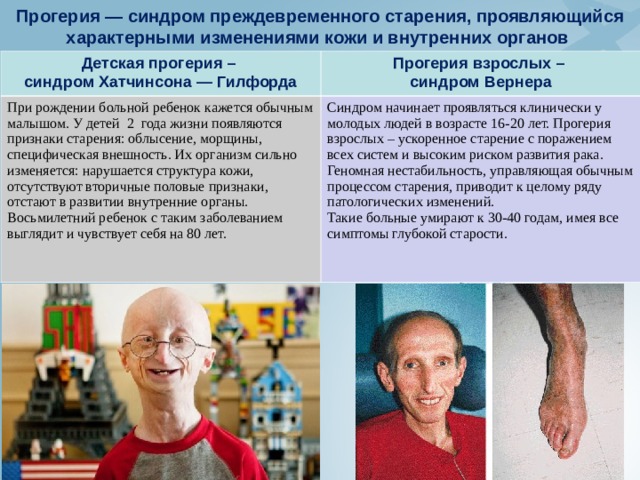
Прогерия — синдром преждевременного старения, проявляющийся характерными изменениями кожи и внутренних органов
Детская прогерия –
синдром Хатчинсона — Гилфорда
Прогерия взрослых –
синдром Вернера
При рождении больной ребенок кажется обычным малышом. У детей 2 года жизни появляются признаки старения: облысение, морщины, специфическая внешность. Их организм сильно изменяется: нарушается структура кожи, отсутствуют вторичные половые признаки, отстают в развитии внутренние органы. Восьмилетний ребенок с таким заболеванием выглядит и чувствует себя на 80 лет.
Синдром начинает проявляться клинически у молодых людей в возрасте 16-20 лет. Прогерия взрослых – ускоренное старение с поражением всех систем и высоким риском развития рака. Геномная нестабильность, управляющая обычным процессом старения, приводит к целому ряду патологических изменений.
Такие больные умирают к 30-40 годам, имея все симптомы глубокой старости.
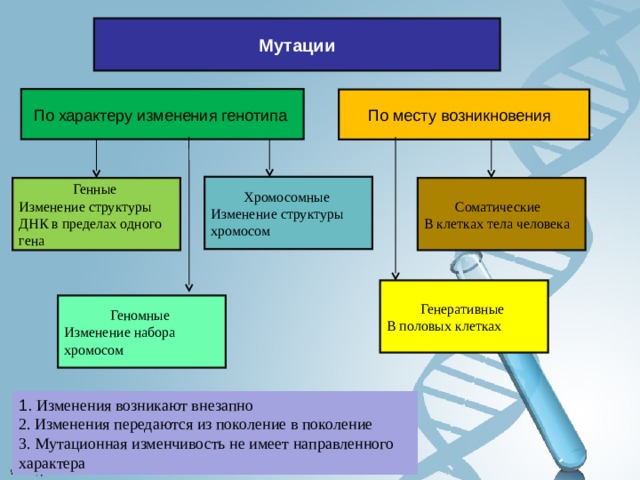
Мутации
По характеру изменения генотипа
По месту возникновения
Хромосомные
Изменение структуры хромосом
Генные
Изменение структуры ДНК в пределах одного гена
Соматические
В клетках тела человека
Генеративные
В половых клетках
Геномные
Изменение набора хромосом
1. Изменения возникают внезапно 2. Изменения передаются из поколение в поколение 3. Мутационная изменчивость не имеет направленного характера

Под первичной профилактикой понимают такие действия, которые должны предупредить рождение больного ребенка. Это реализуется через планирование деторождения путем выбора оптимального репродуктивного возраста, который для женщин составляет 21-35 лет (более ранние и поздние беременности увеличивают вероятность рождения ребенка с врожденной патологией и хромосомными болезнями), и отказа от деторождения в случаях высокого риска наследственной и врожденной патологии (в том числе при браках с кровными родственниками и гетерозиготными носителями патологического гена).

Источники информации
- https://ustamivrachey.ru/genetika/geneticheskie-zabolevaniya
- https://fb.ru/article/291681/geneticheskie-zabolevaniya-peredayuschiesya-po-nasledstvu-mediko-geneticheskoe-obsledovanie
- https://www.krasotaimedicina.ru/diseases/children/shereszewski-turner
- https://fb.ru/article/353754/tomas-hant-morgan-biografiya-vklad-v-biologiyu
- https://fishki.net/1502865-10-samyh-redkih-geneticheskih-zabolevanij.html?sign=224117469314374%2C628925176263087
- https://www.krasotaimedicina.ru/diseases/genetic/Sotos-syndrome
- Бочков Н.П. Клиническая генетика : Учебник / - 3-е изд., испр. и доп. - М. : ГЭОТАР-Медиа, 2004. - 480 с.
- Назаренко С.А. Изменчивость хромосом и развитие человека. - Томск: Изд-во Томского государственного университета, 2008. -200 с.









 Факторы риска возникновении наследственных заболеваний человека Мастер-класс (9.03 MB)
Факторы риска возникновении наследственных заболеваний человека Мастер-класс (9.03 MB)
 0
0 1076
1076 16
16 Нравится
0
Нравится
0


